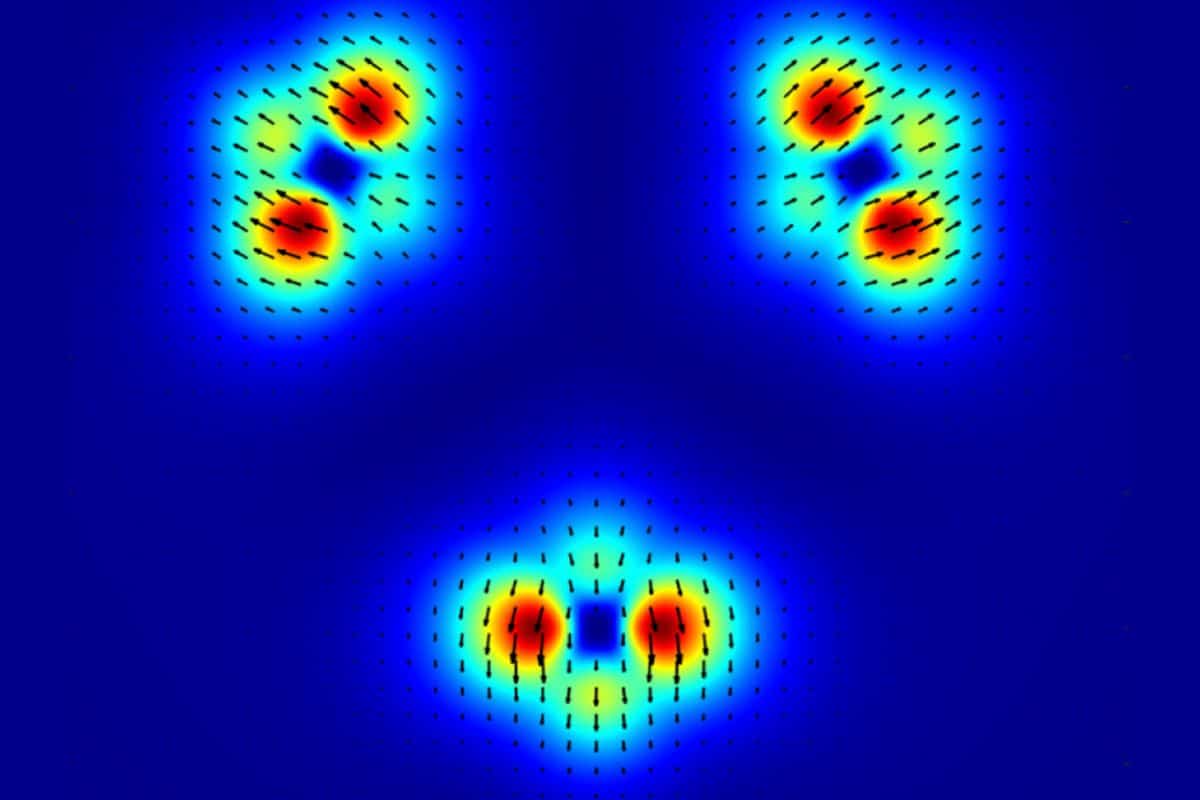
A research team from Cnr Nano, Università di Torino, and National University of Singapore presents an improved approach to calculating magnetic properties of materials using density functional theory (DFT). Published in Physical Review Letters, the research addresses persistent issues in DFT approximations by ensuring they follow a generalized form of the gauge principle. This advancement improves the description of both collinear and non-collinear magnetizations.
Challenges in magnetic property calculations
DFT is a powerful method for modeling complex quantum systems, widely used to predict the electronic structure, and hence many properties, of materials. However, calculating magnetic properties remains challenging due to the intricate nature of magnetic configurations. Improving these calculations is crucial for developing materials with specific magnetic behaviors. In this work, Stefano Pittalis from Cnr Nano and colleagues present a novel approach that resolves some of the most challenging issues with modern DFT approximations as relative to the calculation of magnetic properties of materials.
Overcoming limitations
The research addresses a key limitation of the SCAN (Strongly Constrained and Appropriately Normed) approximation — a widely used DFT method known for its accuracy, but prone to overestimating magnetization. The team developed a refined version of SCAN that corrects this issue, enhancing its reliability in describing ferromagnetic, antiferromagnetic, and non-collinear magnetic states.
A Generalized Approach and results implications
“Instead of tailoring the method to specific magnetic configurations, a key step in our work was to focus on the underlying physical laws that govern all possible magnetic solutions, achieving a more general and reliable approach”, says Stefano Pittalis.
This innovation could support the search for new materials in fields like spintronics, which explores materials for advanced memory and logic devices.
The study combined advanced quantum mechanics, analytical derivations, and software implementation using the Crystal code. The collaboration involved Stefano Pittalis from Cnr Nano in Modena, Jacques K. Desmarais and Alessandro Erba from the Chemistry Department of the University of Turin, and Giovanni Vignale from the Institute for Functional Intelligent Materials at the National University of Singapore. Future work will extend this research to investigate magnetic properties in materials under non-equilibrium conditions.
Original paper: Meta-Generalized Gradient Approximation Made Magnetic, Jacques K. Desmarais, Alessandro Erba, Giovanni Vignale, and Stefano Pittalis, Phys. Rev. Lett. 134, 106402. https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.134.106402
[image: Magnetization of the Cr₃ molecule using the non-collinear SCAN functional]


